- Home
- MCQs
- Cases
- Flashcards
- Illustrations
- E-Text access
- Your feedback
- Become a reviewer
- More student books
- Student apps
- Join an e-mail list

"My baby has a high fever and a bad cough."
Lee, a six-month-old infant, is brought to the pediatric emergency room (ER) by his mother at about 8 AM on a Saturday. She tells you that Lee was well until two days ago, when he developed a cough and seemed lethargic. At that time, she called her family physician. He advised her that it was probably a cold, to keep him well hydrated, and to administer one baby acetaminophen every 6 h if Lee develops a fever. Lee slept poorly that night and the next day was noted to have a low-grade fever of about 38°C. Since he seemed to be getting worse, the family physician was called again, but he continued to think Lee merely had a cold and should be treated symptomatically. Over the course of the day Lee's cough became more frequent, and by the evening he was breathing more rapidly than usual (tachypneic). These symptoms became more intense during the night and prompted the mother's visit to the ER with the baby.
Your physical examination of Lee discloses a child of appropriate length and weight (normal growth) who has a normal overall appearance, that is, no dysmorphic facial features or bone abnormalities. He has an elevated temperature of 38.5°C and obvious respiratory symptoms (coughing and rapid breathing) but no meningeal signs such as neck rigidity or inconsolable irritability. Auscultation of his chest reveals coarse breath sounds but no bronchospasm or evidence of pulmonary consolidation. Examination of the pharynx reveals no evidence of pharyngitis or tonsillitis (inflammation of the phyarynx and tonsils); in fact, the tonsils seem reduced in size.
You surmise that the patient has a pulmonary infection (probably a bacterial pneumonia) and have him admitted to the pediatric ICU (PICU). You also order a chest X ray, blood cultures, and a complete blood count (CBC).
The chest X-ray result is available shortly after admission to the PICU and reveals the presence of bilateral perihilar infiltration. A complete blood count shows an increased white blood cell (WBC) count (15,500 cells/µL, reference range < 12,000 cells/µL) comprised of 72% neutrophils, 7% monocytes, and 20% lymphocytes. Although this differential is compatible with the presence of an infection, you note that the total white count is lower than you expected for a bacterial pneumonia. In any case, you start the patient on IV cefotaxine, a broad-spectrum third-generation cephalosporine antibiotic to treat what you consider is the likely cause of his pneumonia, a bacterial pathogen. You also order a lung computer-aided tomography (CAT) scan to obtain an objective baseline for the extent of the infection.
Over the next several days you return frequently to the PICU to check on Lee's condition. Over this period Lee's respiratory status remains stable but does not improve despite the antibiotic therapy. He continues to be febrile and to be tachypneic. Pulse oxymeter readings show periodic episodes of oxygen desaturation (< 92%) that require treatment with an oxygen mask. The blood cultures have proved negative, so you know neither the identity of the organism nor its sensitivity to antibiotics. Thus, you cannot be sure his infection is being adequately treated and, in fact, his lack of more rapid clinical improvement suggests otherwise. On the morning of the fourth day after admission, Lee's status seems to have deteriorated; his breathing is more labored and oxygen desaturation episodes are more prolonged and frequent. Indeed, a repeat CAT scan shows more extensive lung involvement. After consultation with a pulmonologist, a bronchoscopy is performed to obtain bronchial lavage fluid and brushings for microscopic examination and culture. This is done without incident. The next morning the laboratory report is returned; to your surprise, Lee has an infection with Pneumocystis jiroveci (formerly called Pneumocystis carinii) a protozoal organism that ordinarily causes infection only in immunocompromised hosts. You immediately change the antibiotic to IV trimethoprim-sulfamethoxazole (Bactrim), a specific and effective therapy for this organism.
The results of this treatment are dramatic. Within 24 h Lee shows noticeable improvement. Over the next several days he defervesces and his pulmonary status returns to normal. After four days of IV Bactrim therapy you switch to oral therapy and two days later Lee is discharged to home with the proviso that he continue on oral Bactrim therapy for another two weeks.
What is unusual about a pneumonia due to Pneumocystis jiroveci (carinii)?
Initial Approach
Recognizing that Lee may have an immunologic disorder, you refer him to an immunologist for further workup. The immunologist starts the workup with a second look at the facts already in hand.
History
First, the history of normal development and lack of disease during Lee's first six months of life speak against SCID, an immunodeficiency that generally declares itself early and is associated with a failure to thrive. This tentative conclusion is corroborated by the CBC, which did not show the presence of a lymphopenia (reduced lymphocyte count). It should be noted, however, that Lee was observed to have a reduced tonsillar mass, possibly indicating some loss of ability to populate peripheral lymphoid compartments.
Re-review of the chest X-ray and CAT scans confirms the presence of a thymic shadow, with a size appropriate for age. Combined with the lack of evidence of hypoparathyroidism, dysmorphic facial features, and cardiac anomalies, the presence of a thymic shadow on radiologic examination eliminates consideration of thymic aplasia (DiGeorge syndrome).
Another major consideration in the differential diagnosis is the presence of a neonatal HIV infection (HIV transmitted to Lee from his mother during birth). This is unlikely because an HIV diagnostic test performed on Lee's mother during childbirth (as required by your state's law) was negative. Nevertheless, to be absolutely certain, Lee is tested for the presence of HIV using a Western blot-based technique to detect HIV protein, in case he lacks the ability to mount an antibody response to this organism. The results of this test are likewise negative, and HIV infection is definitively ruled out.
The immunologist also elicits a family history from the mother. He learns that Lee has a maternal uncle (his mother's brother) who died mysteriously at an early age due to pneumonia. This suggests that Lee may have an X-linked disease.
The conclusions drawn from the above review and family history are very valuable but do not identify a specific immunodeficiency state. A more extensive workup is therefore initiated. Although the Pneumocystis infection is drawing attention to the T cells or cell-mediated arm of the immune response, the status of his humoral immunity is also investigated.
Testing and Interpretations
As described in Cases 1 on XLA and the introduction to this unit, an orderly approach to the investigation of the capability of the immune system must be pursued, keeping in mind the stages of development of mature immune cells and the interactions that must take place among them to mount an effective immune response.
Lymphocyte Subset Analysis
Quantitation of peripheral lymphoid subsets by an initial flow cytometry study reveal normal percentages and absolute numbers of CD4+ T cells, CD8+ T cells, B cells, and natural killer (NK) cells. Additional studies are performed in a specialty laboratory that does a more extensive panel of CD markers. Analysis of the CD4+ cell subsets reveals that the percentage of CD4+ cells in the CD4+/CD45RA+ (naive) CD4 T-cell subset is high and, correspondingly, the percentage in the CD4+/CD45RO+ (mature) CD4 T-cell subset is low even if one considers the fact that the ratio of CD45RA+ cells to CD45RO+ cells is high in young children as compared to adults. In addition, while Lee has a normal number of B cells, virtually none bear surface IgG or IgA.
These findings suggest a defect in T- and B-cell maturation at a point governed by antigen exposure.
Immunoglobulin Levels
| Results | Reference Range | |
|---|---|---|
| IgM | 407 mg/dL | 65 ± 25 mg/dL |
| IgG | < 50 mg/dL | 923 ± 256 mg/dL |
| IgA | < 5 mg/dL | 124±45 mg/dL |
How do the results obtained thus far differ from what would be found in studies of patients with XLA, SCID, and common variable immunodeficiency disease (CVID)?
Proliferation Studies
Culture of Lee's cells with nonspecific mitogens, which induce proliferation of T cells (phytohemmaglutinin and concanavalin A) and B cells (pokeweed mitogen) show normal proliferative ability (see Case 1).
In Vitro Immunoglobulin Production
The ability of each cell type to function in the steps required to produce immunoglobulin is next tested.
The B Cells on Their Own
Lee's peripheral blood (circulating) B cells are isolated and cultured in vitro with a CD40L trimer (plus IL-4 and IL-10) to produce antibody in conditions circumventing T-cell dependence. CD40L (CD154) is a molecule appearing on activated T cells that interacts with CD40 on B cells and induces B-cell maturation and class switching; thus, soluble CD40L trimer mimics or replaces the stimulatory activity of T cells. The culture fluid is then assayed for the presence of secreted immunoglobulins.
Result
Lee's B cells, stimulated under these culture conditions, produced normal amounts of IgM as well as normal amounts of IgG and IgA.
This provided solid evidence that his B-cell responses were intrinsically normal and that the serum Ig levels did not reflect an intrinsic B-cell abnormality.
T-B Cell Interaction
In concomitant studies, Lee's T cells are cultured with control B cells and his B cells are cultured with control T cells under conditions to induce polyclonal T-cell stimulation (surface-bound anti-CD3 and soluble anti-CD28). T-cell stimulation leads to the expression of CD40L and cytokine production, and the activated T cell should be capable of inducing B-cell stimulation and isotype switching to IgG and IgA production.
Result
Lee's T cells, stimulated by anti-CD3/anti-CD28, fail to induce normal B cells to produce IgG or IgA, whereas control T cells demonstrate this capacity; on the other hand, control T cells, stimulated by anti-CD3/anti-CD28, induce Lee's B cells to produce IgG and IgA.
These results suggest that Lee's T cells have reduced "helper T-cell activity", that is he has basic T-cell defects, whereas his B cells are normal. Thus his T cells are unable to interact with B cells to induce isotype class switching. This may be the result of a defect in CD40L expression and/or function or a defect in cytokine production.
Cell-Mediated Immunity Studies
Lee's clinical presentation (infection with an opportunistic pathogen) and immunologic workup thus far suggest an underlying T-cell abnormality in spite of the fact that Lee appears to have a B-cell deficiency as manifested by low IgG and IgA levels. The immunologist places particular importance on the T-B-cell interaction studies described above which, given that Lee's B cells are capable of producing immunoglobulin, point to a T-cell defect rather than a B-cell defect. One type of immunodeficiency that fits this pattern is hyper-IgM syndrome, an immunodeficiency that may be caused by failure to express CD40L (CD154) on the surface of activated T cells.
CD40L-CD40 also mediates interactions between T cells and antigen-presenting cells (APCs; monocytes or dendritic cells) to produce cytokines. The absence of effective interaction between T cells and macrophages would be expected to manifest in defective cell-mediated immune responses. To investigate this possibility, the immunologist conducts further T-cell studies that evaluate both in vivo and in vitro T-cell function and T-cell/APC interactions.
Delayed-Type Hypersensitivity Test
These "in vivo" studies consist of intradermal skin tests with tetanus toxoid and diphtheria antigens. Both are antigens to which Lee has already been exposed as part of his normal vaccination schedule. The skin test sites are "read" at 48 h for the presence of induration and erythema to determine if Lee can mount a delayed-type hypersensitivity (DTH) response.
Result
The test proves negative in that neither of these antigens elicits a significant inflammatory response.
Since the DTH reaction depends on T-cell stimulation of APCs via CD40L to produce cytokines (normally resulting in induration and erythrema), the absence of response in these studies is compatible with a CD40L defect. This test of in vivo T-cell immunity also predicts that Lee will unfortunately have further difficulties with opportunistic infections.
In Vitro Activation and Monocyte Cytokine Production: An Investigation of Cell Communication
Lee's peripheral blood mononuclear cells are cultured either with anti-CD3 (an antibody that acts as a T-cell mitogen as noted above) or Staphylococcus aureus Cowan strain 1 (SAC) plus interferon-γ (IFN-γ). The latter combination acts as a direct stimulant of APCs, which, in peripheral blood, consists mainly of monocytes. This time the culture fluid is assayed for the cellular production of secreted cytokines (see Unit V). T cells, when activated by the anti-CD3, normally express CD40L (CD154), which then interacts with CD40 on the monocyte membrane, inducing the monocytes to produce inflammatory cytokines such as interleukin-12 (IL-12) and tumor necrosis factor (TNF)-α. On the other hand, SAC plus IFN-γ stimulates monocytes directly and results in production of IL-12 and TNF-α in the absence of T cells.
Result
Lee's peripheral monocytes produce greatly reduced amounts of IL-12 and TNF-α when they are stimulated by his T cells (cultured with anti-CD3). In contrast, his monocytes produce normal amounts of these cytokines when they are directly stimulated with SAC plus IFN-γ.
Lee's - cells do not stimulate monocytes to produce cytokines, but his monocytes are intrinsically intact. This is likely the result of defective CD40L expression on the T-cells. However, a defect in IFN-γ production by the T-cells could also be the culprit; therefore that possibility is tested next.
FN-γ Production
Lee's mononuclear cells are cultured with anti-CD3, but this time CD40L stimulation of the monocytes is "artificially" provided by addition of CD40L trimer, the soluble form of CD40L that can stimulate monocytes via CD40 in the absence of T cells. The CD40L trimer was used for a similar purpose in B-cell cultures (see above). T cells in cultures with anti-CD3 and activated monocytes should produce a cytokine such as IFN-γ as a result of stimulation by the anti-CD3 and the helper cytokines produced by the CD40L-stimulated monocytes.
Result
Whereas Lee's T cells produced greatly decreased amounts of IFN-γ in the absence of CD40L trimer, they produced normal amounts in its presence. The control T cells, on the other hand, produced IFN-γ in the presence and absence of CD40L trimer.
These data thus offer excellent functional evidence that Lee's T cells are functionally deficient either because they do not express CD40L or because they express an inactive CD40L, rather being unable to produce IFN-γ.
Detection of the Molecule
At this point the immunologist closes in on the diagnosis of X-linked hyper-IgM syndrome (XHIGM; HIGM-I) by performing flow cytometry studies using fluorochrome-labeled anti-CD40L. These studies indeed demonstrate that Lee's cells do not express CD40L. The immunologist also sends a sample of Lee's DNA to a sequencing facility for sequencing of the CD40L gene. The results show that Lee's CD40L gene has a mutation in the extracellular domain of the gene that results in a truncation of this domain and thus low expression of CD40L on the T-cell surface. The diagnosis of XHIGM is now secure. Given the family history of a maternal uncle with an illness that could have been caused by HIGM, Lee is assumed to have inherited this mutated X-linked gene rather than to have a new mutation. This was subsequently verified by sequencing of Lee's mother's DNA, which showed that the mother carries the mutated gene on one X chromosome.
What is the explanation for the initial observations that Lee's tonsils are underdeveloped and that his peripheral helper T cells are skewed toward the naive (CD45RA) subset rather than the memory (CD45RO) subset?
Lee is placed on monthly intravenous immunoglobulin (IVIG) infusions of 20 mg/kg to bring his "trough" (low-point) IgG level above 500 mg/dL, the target IgG level for immunodeficiency patients who cannot produce normal amounts of IgG. In addition, he is placed on prophylactic Bactrim therapy to prevent recurrence of Pneumocystis infection.
At the age of three years, Lee developed a neutropenia (2000 neutrophils/μL versus the reference level of 5000 neutrophils/μL), a known concomitant of XHIGM syndrome. At age six years, he was placed on granulocyte colony-stimulating factor (G-CSF) therapy and partial amelioration of the neutropenia was achieved. This neutropenia was associated with recurrent apthous mouth ulcers, which tended to improve when neutrophil levels increased. Extensive attempts to culture an organism from these ulcers were unsuccessful.
At age eight, Lee suddenly developed mild right-sided weakness. Unfortunately, this symptom was not investigated until three months later, when he had a grand mal seizure. At that point a magnetic resonance imaging (MRI) examination of the brain revealed multiple punched-out lesions with enhancement. This picture is typical of toxoplasmosis, a known complication of XHIGM. Lee was therefore put on long-term pyrimethamine-sulfadiazine therapy that had an immediate effect as evidenced by the decrease in the enhancement in the MRI and had the long-term effect of preventing progression of the toxoplasmosis.
As will be detailed in a later section, the clinical syndrome of hyper-IgM has several possible pathophysiologic bases. Here we will discuss the clinical features of XHIGM syndrome. Although the name "hyper-IgM" places the emphasis on the inability to isotype switch to IgG and IgA immunoglobulin production, the infectious complications of XHIGM remind one that this is a deficiency in an interaction (CD40-CD40L) important in both humoral and cell-mediated immune responses. Thus there is overlap in its clinical features with those of some forms of SCID and T-cell immunodeficiency in that these patients have infections not normally found in conditions such as CVID (see below), a disorder with usually intact cell-mediated immunity.
Pulmonary Problems
Since patients with all forms of HIGM syndrome have increased or normal amounts of IgM levels but decreased IgG and IgA levels, it is not surprising that they are susceptible to infection with extracellular pathogens, similar to patients with CVID. Indeed, upper and lower respiratory infections by extracellular pathogens are the most common problems of hyper-IgM patients. In the absence of IVIG replacement therapy, bronchiectasis is likely to result as a consequence of recurrences. However, respiratory infection in X-linked hyper-IgM syndrome is also not infrequently due to opportunistic organisms (unlike CVID), and in over 40% of patients in whom pneumonia marked the onset of disease in the first year of life, Pneumocystis jiroveci was the cause of infection. Other opportunistic organisms causing infection in XHIGM patients include cytomegalovirus (CMV), cryptococcus, and atypical mycobacterial infection. These are the consequence of the inadequate interaction between T cells and macrophages via CD40-CD40L.
Gastrointestinal Problems
XHIGM patients frequently have diarrhea, and in 20-30% of cases, this can have a chronic course, associated with malabsorption and failure to thrive (again similar to what occurs in CVID). At times, the diarrhea results from opportunistic infection, such as with cryptosporidia or giardia, but in other instances it appears to be noninfectious in nature. In these latter cases, the patients may have intestinal nodular lymphoid hyperplasia and villous atrophy, suggesting the presence of an autoimmune process involving the GI tract.
Liver Problems
Severe liver problems occur in almost 20% of XHIGM patients. These can take the form of sclerosing cholangitis (frequently due to cryptosporidiosis) as well as infection with hepatitis B and hepatitis C or even CMV. The hepatic infections can lead to cirrhosis and liver decompensation, which may necessitate a liver transplant. Historically, hepatitis in hyper-IgM syndrome has been associated with exposure to contaminated blood products, thus, IVIG used for replacement therapy must come from an unimpeachable source. Another complication of chronic hepatitis in XHIGM patients is hepatocellular carcinoma, which usually has a fatal outcome.
Hematologic Manifestations
The main hematologic problems in XHIGM patients are anemia and neutropenia. The former occurs in about one-third of patients and is usually due to iron deficiency; however, the anemia can occasionally be due to bone marrow failure (aplastic anemia) or peripheral lysis of the red cells (hemolytic anemia). Neutropenia, the more common hematologic abnormality, occurs in almost 70% of patients. It takes the form of a block in myeloid cell development, occurring at the promyelocytic-myelocytic stage. In some cases, the neutropenia is chronic and in other cases it is cyclic. It is frequently associated with gingivitis and perioral ulceration.
Neoplasia in XHIGM patients
Visceral neoplasms involving the gall bladder, liver, and pancreas have been reported, but this is not a frequent phenomenon. Lymphomas, often increased in other immunodeficiency states, are not associated with XHIGM, possibly because patients do not live long enough to develop this complication.
Miscellaneous Problems
A degenerative encephalopathy is not infrequently seen in XHIGM patients and is usually due to infection with any of several opportunistic pathogens, including Cryptococcus, toxoplasmosis, CMV, Echo virus, and Mycobacterium bovis. Because some of these infections are treatable, vigorous search for a causative agent should be done at the onset of CNS symptoms. Once degenerative changes occur, they can rarely be completely reversed.
Finally, secondary arthritis occurs in a small group of patients. This is usually associated with neutropenia and is presumably due to infection, but in most cases a causative agent cannot be identified.
The maturation of B cells occurs in two phases. The first is an antigen-independent phase occurring in the fetal liver and bone marrow and the second is an antigen-dependent phase that occurs in lymphoid peripheral tissues such as the Peyer's patches, the spleen, and the lymph nodes. The steps necessary to advance the B cell from the progenitor stage to a naive mature B cell leaving the bone marrow were reviewed in Case 1 on XLA. In that first phase, the "germline" B-cell repertoire is established in individual B cells by rearrangement of both the heavy- and light-chain immunoglobulin genes. These rearrangements result in the formation of a VDJ segment from the variable segments, diversity segments, and joining segments (V, D, and J segments, respectively) as a consequence of a random recombination process. At the conclusion of these molecular events, the Ig heavy-chain gene consists of a "recombined" VDJ segment that determines antigen specificity. This segment sits upstream of a string of heavy-chain constant-region segments, the first of which are the Cμ and Cδ segments. The cell is now able to produce IgM and IgD antibodies.
In the second (antigen-dependent) stage of development, which occurs in the periphery, B cells undergo proliferation. At the same time, their Ig genes may undergo further changes, that of somatic hypermutation in the variable region (resulting in affinity maturation) and that of isotype class switch rearrangement. Somatic hypermutation is the result of point mutations in the VDJ segments, which lead to subtle changes in the affinity of the immunoglobulin for antigen. This process allows for the generation of cells producing high-affinity antibodies. Isotype class switch rearrangement results in a substitution of the constant-region portion of the heavy-chain molecule. Thus, the VDJ becomes associated with various downstream "C-region" heavy-chain genes such as γ, α, or ε C regions. In this way cells producing the various classes and subclasses of immunoglobulins are formed and combined with the hypermutated variable portion; these antibodies thus produced have a generally higher affinity for antigen than IgM.
In patients with hyper-IgM syndrome, the first phase of B-cell maturation, the antigen-independent phase in the bone marrow, is intact. It is in the second, antigen-dependent stage in the periphery that progression is halted (Fig. 4.1).
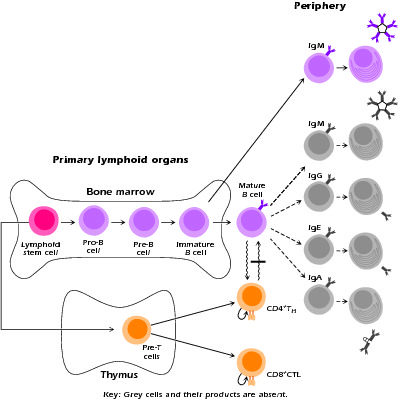
Figure 4.1 Sites of defective lymphopoietic development associated with primary immunodeficiency syndromes. Developing and mature lymphoid populations remaining in patients with hyper-IgM syndrome. Gray cells denote absent populations. Bar denotes block in interaction.
The Structural Basis of Class Switching
What is the molecular basis of the process by which B cells switch from the production of IgM to IgG or IgA; in other words how do they undergo Ig class switch differentiation?
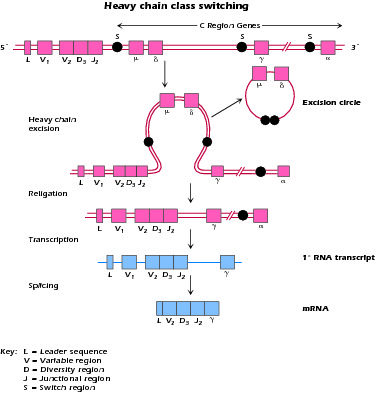
Figure 4.2 Mechanism of class switching in Ig synthesis; S = switch region, upstream of each heavy-chain constant region. Switching requires T cell interaction.
What signals the B cell to undergo isotype switching?
The "Purpose" of the Different Immunoglobulin Constant Regions
Whereas the variable region governs the antigen-binding properties of the immunoglobulin molecule, the constant region determines its biologic properties. This region is required for recruiting other cells via their Fc receptors, binding complement, and specifically transporting the immunoglobulin molecule via active transport mechanisms to other sites. Macrophages and neutrophils, the body's phagocytic cells, have Fc receptors for IgG1 and IgG3, whereas high-affinity FcRε are found on mast cells, basophils, and eosinophils, the cells containing mediators needed for response to parasitic infections. Complement, which may directly destroy pathogens or activate phagocytes, is bound most efficiently by IgM, IgG3, and IgG1. IgG1 and IgG3 cross the placenta and make their way into the fetal circulation. IgA is specifically and actively transported across the epithelial cell into mucous secretions, tears, and milk (Table 4.1).
Table 4.1 Most Important Features of Immunoglobulin Isotopes
| Characteristic | IgG | IgA | IgM | IgD | IgE |
|---|---|---|---|---|---|
| Placental passage | ++ | — | — | — | — |
| Presence in secretion | — | ++ | — | — | — |
| Presence in milk | + | + | 0 to trace | — | — |
| Activation of complement | + | — | +++ | — | — |
| Binding to Fc receptors on macrophages, polymorphonuclear cells, and NKa cell | ++ | — | — | — | — |
| Relative agglutinating capacity | + | ++ | +++ | — | — |
| Antiviral activity | +++ | +++ | + | — | — |
| Antibacterial activity (Gram negative) | +++ | ++ (with lysozyme) | +++ (with complement) | — | — |
| Antioxin activity | +++ | — | — | — | — |
| Allergic activity | — | — | — | — | +++ |
| IgG1 | IgG2 | IgG3 | IgG4 | ||
| Complement binding | + | + | +++ | — | |
| Placental passage | ++ | ± | ++ | ++ | |
| Placental passage | +++ | + | +++ | ± |
a. Natural killer.
As its name implies, HIGM syndrome is defined by the fact that patients have B cells that produce IgM but not IgG, IgA, or IgE. Thus, they have a defect in class switch recombination. As is evident from the discussion above, class switch recombination is a multistep process and thus the HIGM syndrome could result from molecular defects affecting any of these steps. As shown in Figure 4.3, five distinct defects leading to the HIGM syndrome have so far been identified. Three of these defects (designated HIGM-I, III, and IV) affect the function of B cells, T cells, and APCs; two of these defects, HIGM-II and V, affect the function of B cells only. The most common form is X-linked HIGM, described in our patient, Lee.
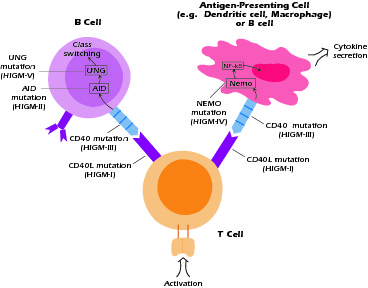
Figure 4.3 Defects in hyper-IgM syndromes. Some defects interfer with T-macrophage interaction as well as T-B interaction. Others interfere with mechanisms of class switching within the B cells.
Molecular Basis of XHIGM, HIGM-I Syndrome
An important clue to the molecular origin of the most prevalent form of hyper-IgM syndrome came from the discovery that class switch rearrangement is an antigen-driven process that occurs in germinal centers of lymphoid follicles. Thus, when it was discovered that interaction between CD40 on APCs and CD40L on T cells was necessary for germinal center formation, investigators considered the possibility that this interaction was important for class switch rearrangement and by extension was somehow involved in hyper-IgM syndrome. This suspicion was heightened by the fact that most patients with HIGM syndrome have lymphoid organs characterized by poorly developed germinal centers. It was ultimately shown that an X-linked form of hyper-IgM syndrome (XHIGM or HIGM-1), representing about 50% of all cases, is due to a mutation of the CD40L gene, resulting in nonexpression of CD40L or expression of a defective CD40L that cannot exert signaling function. Figure 4.4 shows a diagram of the CD40L molecule and the approximate locations in the molecule of mutations associated with XHIGM. Most of the mutations occur in the gene segment encoding the extracellular domain of the molecule and result in stop codons or base substitutions that cause loss of expression of CD40L on the cell surface or a CD40L molecule that cannot signal via CD40.
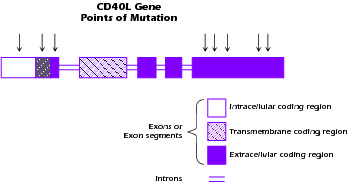
Figure 4.4 Structure of CD40L (CD154) gene. Arrows denote positions of mutations.
The CD40L molecule is a member of the TNF-α receptor family of molecules; its interaction with CD40 results in activation of important signaling pathways that affect the function of the CD40-expressing cells, B cells, and APCs, particularly dendritic cells. As noted above, CD40 signaling in the B cell is necessary for B-cell proliferation, germinal center formation, and class switch rearrangement. In dendritic cells, CD40 signaling is important in the production of key cytokines such as IL-12 and in the expression of costimulatory molecules such as CD80 and CD86. Thus, as shown in Figure 4.5, the consequences of a mutation of CD40L include not only the functional abnormalities in B cells that are the hallmark of the disease, but also T-cell abnormalities due to defective interactions with APCs. The latter is illustrated in Figure 4.5C, which shows the sequential activation of T cells leading to expression of CD40L and the signaling of dendritic cells to produce IL-12. Both the B-cell and APC "defects" are actually secondary to the absence of signal from the real culprit, the T cell that lacks functional CD40L. The main immunologic consequence of the inadequate APC-T cell interaction is decreased IL-12 production, leading in turn to decreased production of IFN-γ by T cells. This explains why hyper-IgM patients with CD40L mutations (XHIGM patients) have increased susceptibility to infection with opportunistic organisms. With the advent of Ig replacement therapy, this defect in XHIGM patients is now the most significant clinical abnormality remaining and is one that explains their clinical course.
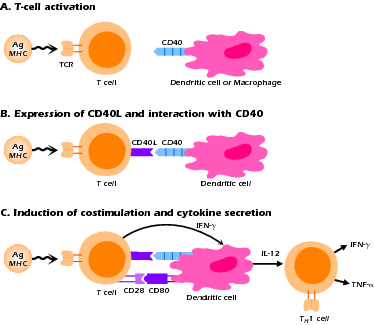
Figure 4.5 Sequence of events in T cell-APC interaction through CD40-CD40L following antigen stimulation.
Interestingly, XHIGM patients with mutations in CD40L have IgM+/IgD+ B cells (unswitched B cells) that express CD27 (a maturation marker) and that exhibit evidence of some somatic hypermutation. These cells may have been generated in non-germinal center areas (e.g., marginal zones) as a result of other interactions, such as those involving OX40/OX40L, another TNF receptor family member. They can probably react with thymic independent (polysaccharide) antigens and thus provide patients some protection against disease. Along these lines, it also has been noted that a substantial fraction of patients produce IgA but not IgG. This may arise from class switching in certain types of B cells that does not require CD40L.
Other Forms of Hyper-IgM Syndrome
In recent years several other forms of hyper-IgM syndrome have been described; they are not due to a CD40L mutation, but like XHIGM they present as a defect in class switch recombination. As discussed below, some of these syndromes are characterized by considerably different clinical patterns than that found in XHIGM.
Hyper-IgM Syndrome due to CD40 Mutations: HIGM-III
Several patients with hyper-IgM syndrome have been reported to have an autosomal recessive form of the disease in which various mutations in the CD40 gene lead to failure to express CD40 on the cell surface. Inasmuch as this abnormality leads to the same immunologic defects as seen in XHIGM (absence of CD40-CD40L interaction), it is not surprising that the clinical pattern found in these patients closely resembles that of XHIGM. This form of hyper-IgM syndrome, which has been found in females (and can potentially occur in males as well), is classified as HIGM-III.
Hyper-IgM Syndrome Due to NEMO Mutations: HIGM-IV
Another form of HIGM syndrome is X linked but has a different molecular defect than XHIGM (HIGM-I). This is known as XHIGM-ED or HIGM-IV. It occurs in association with hypohydrotic ectodermal dysplasia (ED) and has been shown to be due to mutation in nuclear factor κB (NF-κB) essential modulator (NEMO). NF-κB is a master transcription factor controlling expression of numerous gene programs and functioning in different cell types. NEMO is a docking molecule for Iκκα and Iκκβ, two phosphorylases involved in the phosphorylation and degradation of IκBa and thus in the activation of NF-κB components; it is also involved in the translocation of these components to the nucleus. As shown in Figure 4.6, interaction of CD40L with CD40 leads to the activation of second messengers that interact with NEMO and initiate the activation and nuclear translocation of NF-κB components. Thus, a mutation in NEMO that results in impairment of its function leads to a defect in CD40L signaling and another form of the HIGM syndrome. It should be noted, however, that intact NEMO function is also necessary for the activation of NF-κB by numerous other cell stimulants, such as Toll-like receptors; this explains the fact that cells of patients with this form of HIGM display abnormal responses to these stimulants as well as CD40L.
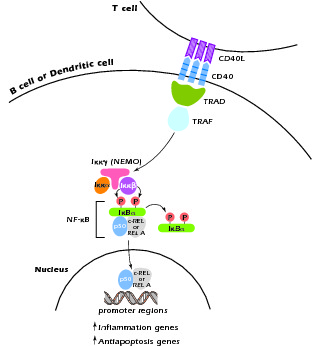
Figure 4.6 CD40L signaling at the cell surface leads to activation of NF-κB with subsequent translocation of its components to the nucleus. The inital step of NF-κB activation involves the interaction of TRAF with NEMO.
The clinical features of this form of hyper-IgM syndrome are thus similar to those with XHIGM due to CD40L deficiency. However, given the ubiquitous nature of NF-κB and its interacting molecules, it is not unexpected that these patients have additional abnormalities. XHIGM-ED is associated with ectodermal dysplasia because the development and function of ectodermal tissue depend on another TNF family receptor that also depends on NEMO for signaling (the DL or ED receptor). The ectodermal abnormalities include inability to sweat (hypohydrosis), teeth malformations (hypodontia), and hairlessness. Susceptibility to infection is confined to high-grade extracellular pathogens rather than including opportunistic pathogens as well but is aggravated by the fact that the patients have thick secretions that impede elimination of organisms from the airways.
Hyper-IgM Due to Activation-Induced Cytidine Deaminase (AID) Mutations: HIGM-II
This fourth form of hyper-IgM syndrome, HIGM-II, which is inherited in an autosomal recessive manner, is caused by a mutation in the gene encoding AID, a molecule essential for both somatic hypermutation and class switching in B cells. As in the case of XHIGM-ED, these patients have normal CD40L and CD40 expression. Because somatic hypermutation is severely impaired in the absence of AID, the B cells of patients with HIGM-AID elaborate only low-affinity IgM antibodies.
Patients with this form of hyper-IgM syndrome differ from patients with other hyper-IgM syndromes in that they have a defect exclusive to their B cells. The CD40-CD40L interactions between T cells and macrophages and T cells and dendritic cells are intact; therefore these patients are not subject to opportunistic infection or other problems related to cell-mediated immunity. In this form of the syndrome, patients do manifest lymphoid hyperplasia and various autoimmune manifestations.
Hyper-IgM Syndrome Due to Uracil-DNA Glycosylase Mutations: HIGM-V
Yet another form of hyper-IgM syndrome has been identified and this one (HIGM-V) is due to mutations in a gene known as uracil-DNA glucosylase (UNG) This enzyme is expressed in activated B cells and operates downstream of AID during the class switch recombination: AID deaminates cytosine to form uracil and then the uracil is removed by UNG. The clinical picture in this hyper-IgM syndrome is similar to that seen due to AID deficiency.
Replacement of IgG by IVIG administration is the main thrust of therapy and, indeed, has resulted in a marked decrease in infection in these patients. In some (but certainly not all) cases, this leads to normalization of elevated IgM levels and even correction of the neutropenia. Adjunctive therapy consists of treatment of the neutropenia with G-CSF, and prophylactic Bactrim therapy for the prevention of Pneumocystis pneumonia in XHIGM patients. Despite these therapies, many patients with XHIGM continue to experience opportunistic infections. Because of this, survival of these patients beyond the second decade has been less than 20%.
On a more fundamental level, several patients have been treated successfully with bone marrow transplantation, usually from a human leukocyte antigen (HLA)-matched sibling. In view of the poor prognosis of XHIGM patients, this therapy should probably be considered more frequently, especially when a matched donor is available.