- Home
- MCQs
- Cases
- Flashcards
- Illustrations
- E-Text access
- Your feedback
- Become a reviewer
- More student books
- Student apps
- Join an e-mail list

"Sam always gets sick; now he has a bad earache and fever."
Sam is a four-year-old boy who is brought to you in the emergency room (ER) by his distraught parents because he has a fever, has been vomiting in the last few hours, and seems slightly disoriented (confused). His mother relates that initially Sam complained of a right-sided earache that was only transiently relieved with acetaminophen. Over the next few days, he developed a fever and became less active and more irritable and did not want to eat. Yesterday he began complaining of a headache "all over his head" and this morning he began vomiting. His mother states that Sam has a history of frequent ear infections (otitis media) with effusion but is concerned that this time it is more than just an earache.
Sam has a generally normal appearance and size but that he has an acute illness marked by fever, withdrawal from his surroundings, and extreme irritability, that is, crying spells unabated by his mother's attempts to calm him down. He has a temperature of 40°C, his skin is hot to the touch, and he appears to be mildly dehydrated (as evidenced by dryness around his mouth and eyes). His throat is injected (red) but without exudate (pus) and his right ear has an opacified tympanic membrane with a loss of the light reflex; his left ear is normal. He has nuchal rigidity (neck stiffness when his head is flexed). You evaluate him for other signs of meningeal irritation and find that he has a positive Kernig's sign (pain when lower leg is extended with his hip flexed at 90°) and a positive Brudzinski's sign (involuntary flexion of the knees and hips when you flex his neck).
His lungs are clear to auscultation and his abdomen has normal bowel sounds, is nondistended, and is nontender. His joints are nontender and mobile
You are alarmed at the evolution of Sam's initial complaint and his neurologic signs. Suspecting that he has an infectious meningitis you immediately perform a lumbar puncture (after checking his eyes for papilledema with an ophthalmoscope) to confirm the diagnosis and to attempt to identify the organism. The cerebrospinal fluid (CSF) is obtained and blood and CSF samples are sent for emergency analysis and to the microbiology lab for culture.
Results obtained quickly are as follows:
| Cerebrospinal fluid: | Result | Reference Range |
|---|---|---|
| Opening pressure | 180 mm Hg | 50-80 mm Hg |
| Appearance | Cloudy | Clear |
| White blood cells (WBC) | 1078/µL | <5/µL |
| Neutrophils | 75% | <25% |
| Red blood cells (RBC) | 0 | 0 |
| Protein | 300 mg/dL | 20-45 mg/dL |
| Glucose | 3 mg/dL | > 50 mg/dL |
| Peripheral WBC count and differential: | Absolute Count | Reference Range |
|---|---|---|
| Total WBC | 24,000/µL | 5500-15,000 |
| Neutrophils 75% | 18,000/µL | 1500-8500 |
| Lymphocytes 19% | 4,560/µL | 2000-8000 |
| Monocytes 5% | 1,200/µL | 300-650 |
| Eosinophils 1% | 240/µL | 100-500 |
In view of the above history and laboratory findings, what is your initial diagnosis and treatment plan?
The next morning, after almost 24 h of intravenous ceftazidine, you walk into Sam's hospital room to find that he is calm, smiling faintly, and beginning to eat. His parents are much relieved and are already asking when Sam can be taken home. The laboratory reports that Streptococcus pneumoniae was isolated from both his blood and CSF but antibiotic sensitivities are not yet available; you therefore continue the ceftazidine therapy. When they become available, you may elect to change the antibiotic on the basis of the sensitivities of the isolated streptococcal organism.
Now that the urgent problem of bacterial meningitis is resolving, you obtain a more complete history from the parents. The mother mentions again that Sam seems to get sick quite often, definitely more often than his playmates. She relates that Sam had skin abscesses at 9 and 10 months of age, three episodes of pneumonia documented by X-ray findings, multiple episodes of diarrhea, and one episode of left knee swelling. His parents take him to the pediatrician on a regular basis.
This history of multiple, severe, and in some cases unusual infections is disturbing and you decide that further investigation is needed. You consider the possibility that Sam has a primary immunodeficiency disorder and refer the family to an allergist/immunologist for more definitive workup once he has recovered from the acute infection.
Three weeks later, Sam has fully recovered from the meningitis and has completed the recommended antibiotic treatment (the streptococcus was sensitive to ceftazidine). At this point he is seen by an allergist/immunologist who confirms that Sam is within the expected range for height and weight and has a normal appearance. On examination, however, he notices that Sam lacks tonsillar tissue, quite an abnormal finding in a 4-year-old child. There is no lymphadenopathy (enlargement of lymph nodes) or hepatosplenomegaly (enlargement of liver and spleen). He also checks Sam's blood count and finds that it has returned to normal.
From questioning the parents, the allergist/immunologist learns that Sam is an only child and that both parents are healthy. The pregnancy was uneventful and Sam was a full-term baby of normal size. The parents do not remember any fevers or infections during the first 6-8 months of his life. In relating the family history, Sam's mother states that her two sisters are healthy but that she had a brother who died at age 18. She recalls that the brother was always sick and had repeated pneumonias resulting in chronic lung disease. Her male cousin died of polio after receiving an oral polio vaccine during childhood. Sam's father's family history is negative for family members with multiple infections.
The allergist/immunologist concludes that Sam's history and the family history are sufficiently suggestive to warrant an immunodeficiency workup.
What information does one seek in such a workup?
Tests may be organized to investigate each arm of the immune system (see the Introduction to this unit and below). In addition, specific assays either provide quantitative information or measure functional activity. In general, the results of quantitative assays are more quickly available. Given the fact that Sam has a sufficiently strong personal and family history to justify this workup, most of these tests are initiated at the outset. As the results are collected and evaluated, it will become obvious that some give predictable results. For teaching purposes, we will present the results of this evaluation arranged according to the immune system being probed.
Serum Immunoglobulin Levels: Quantitative Assay
Blood is drawn from Sam for determination of serum immunoglobulin levels. These are measured by a spectrometric assay called nephelometry, with the exception of IgE, which, because of its normally low level, requires a more sensitive method: radioimmunoassay (RIA) or enzyme linked immunosorbent assay ELISA (see unit V for detailed assay descriptions).
Result
| Reference Range | ||
|---|---|---|
| IgG | 140 mg/dL | 923±256 mg/dL |
| IgM | 32 mg/dL | 65±25 mg/dL |
| IgA | 48 mg/dL | 124±45 mg/dL |
| IgE | 1.5 µg/dL | 2.0-10 µg/dL |
What information do you obtain from overall immunoglobulin levels?
Sam shows abnormally low levels of all the immunoglobulin subclasses.
Specific Antibody Responses to Antigenic Challenge: Functional Assays
Retrospective
Studies of response to previous challenge. Blood is drawn for the determination of measles, mumps, rubella, and varicella serum antibody titers with the knowledge that Sam had been administered vaccines for these organisms at approximately 12 months of age. These antibodies are measured by ELISA using the specific antigens. This allows detection of low levels of antibody, as one would expect in response to a specific antigen, and allows titers to be obtained.
Result
Titers to all four organisms come back as "undetectable" or at the lowest limit of detection.
Prospective
Response to a new challenge. In a more definitive test of ability to mount an antibody response, Sam is administered a standard dose of diphtheria and tetanus (DT) antigens as well as pneumococcal antigens to test his capacity to mount an antibody response to protein (DT) or to polysaccharide (pneumococcal) antigens [Coico and Sunshine (Chapter 12) for description of T-dependent and T-independent antigens]. Blood is drawn just prior to antigen administration and again at two and four weeks after immunization. Serum antibody titers to each specific antigen are measured at the three time points. Although Sam would already have been given three doses of DT as part of the routine vaccination schedule, a normal individual will exhibit at least a 5- to 10-fold rise in titer to DT following this immunization protocol and a 4- to 5-fold rise in titer to one or more of the pneumococcal subtypes. The data are obtained about two weeks after the submission of the sera.
Result
Sam is unable to mount a significant antibody response to either DT or pneumococcal antigens.
Conclusion of Humoral Immunity Studies
The low "across-the-board" Ig levels and poor antibody responses point to a severe deficiency in B-cell function. However, from this information one cannot state definitively that this is the only immune defect present or that the B cells are the culprits.
Analysis of Lymphocyte Subsets in the Peripheral Blood: Quantitative Studies by Flow Cytometry
Sam's peripheral blood cells are stained with fluorochrome-bound antibodies recognizing CD markers characteristic of particular lymphocyte subsets. The number of cells staining with each antibody is quantitated using a flow cytometer (see Unit V).
Result
| Reference Range | Absolute Count | ||
|---|---|---|---|
| CD3 (pan T-cell marker) | 85% | 55-82% | 1755/µL |
| CD4 (marker of helper T cell) | 55% | 33-57% | 1053/µL |
| CD8 (marker of cytotoxic T cell) | 31% | 8-34% | 725/µL |
| CD19 (B-cell marker) | 0% | 6-22% | 0 |
| CD16/56 (NK cell markers) | 15% | 6-31% | 350/µL |
Combining the Data
The lymphocyte subset quantification studies suggest that Sam's inability to mount an antibody response is due to the absence of mature B cells.
In Vitro Proliferation Studies: Nonspecific Functional Assays
Sam's cells are cultured with various stimulators for three days, after which their level of proliferation is quantitated by their ability to incorporate tritium-labeled thymidine. Agents that stimulate T-cell proliferation include the mitogens phytohemagglutinin (PHA) and concanavalin A (ConA) and the T-cell-specific antibody combination anti-CD3 and anti-CD28. B cells are stimulated to proliferate by incubation with pokeweed mitogen (PWM), Staphylococcus aureus Cowen type I, and the B-cell specific antibodies anti-µ heavy chain or anti-CD40.
Sam's cells are cultured with the anti-CD3/anti-CD28, which is designed to mimic the manner in which T cells are normally stimulated, and with anti-CD40, which is designed to mimic the normal interaction between T helper cells and B cells.
Result
Anti-CD3/anti-CD28 stimulation: the proliferation response of Sam's T cells is normal as compared to simultaneously run control cells.
Anti-CD40 stimulation: the proliferation of Sam's cells is significantly decreased as compared to simultaneously run control cells.
These phenotyping and proliferation studies reveal the presence of a B-cell defect, corroborating the humoral immunity studies which focused on B cells. Given the fact that Sam has no detectable levels of B cells in the peripheral blood, stimulation with B-cell specific mitogens or anti-CD40 was expected to be undetectable. It is also not surprising that Sam is unable to mount an antibody response given the absence of mature B cells (and tonsillar tissue). The results additionally demonstrate that Sam has normal T- and NK cell numbers and that Sam's T cells can mount a normal proliferative response to stimulation via the specific receptors that would normally be involved in a T-B collaboration. Thus, his T cells appear to be normal.
Delayed-Type Hypersensitivity (DTH) Test: In Vivo T-Cell Functional Assay
An antigen to which Sam has been previously exposed is injected intracutaneously on the forearm and development of redness and induration (swelling) is evaluated at 48 h. Since Sam has been immunized with tetanus toxoid in the past, tetanus toxoid is injected intracutaneously in the forearm area.
Result
At 48 h, a 1 cm area of redness and swelling is noted.
DTH is a classic test for cell-mediated immunity but in fact only measures the interaction between T helper cells and APCs (along with nonspecific vascular changes, etc.). Additional tests of cell-mediated immunity include studies of cytotoxic T-cell function; however since all evidence, both clinical and laboratory, suggests that this arm of Sam's immune system is intact, they will not be performed in this case (see differential diagnosis section below and Unit V).
For completeness, Sam's blood is also sent to the laboratory for the determination of complement levels and the ability of his white cells to generate reactive oxygen species or hydrogen peroxide. The latter is a function of phagocytes and is impaired in chronic granulomatous disease.
Complement Studies: Quantitative Assay of Complement Components
Result
| Reference Range | ||
|---|---|---|
| CH50 | 180 µg/ml | 150-310 µg/ml |
| C3 | 95 mg/dL | 85-201 mg/dL |
| C4 | 30 mg/dL | 16-47 mg/dL |
Phagocytic Cells: Functional Assays of Neutrophils
Nitroblue Tetrazolium (NBT) Test or Dihyrorhodamine (DHR) Assay
In the NBT Test, yellow NBT is converted to blue/violet formazan within normal neutrophils due to reduction of NBT by O-2, thus measuring their ability to generate reactive oxygen species. In the DHR Assay, lack of DHR oxidation and production of red rhodamine by superoxide oxidation is measured by flow cytometry.
Result
Sam's cells exhibit blue/violet crystals following incubation with NBT.
Flow Cytometric Analysis of PMA-Stimulated Neutrophils Loaded with Dihydrorhodamine
When dye is oxidized by H2O2, it exhibits increased fluorescence, demonstrating the neutrophils' ability to generate H2O2 when stimulated.
Result
Sam's cells exhibit normal flow patterns.
Complement levels and neutrophil functions are normal.
Based on the above results, what is your overall conclusion concerning the functioning of Sam's immune system and location of possible defect?
Why was Sam healthy until the age of six months?
Based on the laboratory results, the allergist/immunologist concludes that Sam has a B-cell disorder. Since this has led to an inability to produce normal levels of immunoglobulins and mount normal antibody responses, he starts Sam on intravenous immunoglobulin (IVIG) therapy administered every four weeks as a replacement for his own inability to produce immunoglobulins. After a single IVIG administration Sam's trough IgG level (the level obtained four weeks after the initial dose and just prior to the second dose) is 400 mg/dL. His trough level after six months of therapy is 550 mg/dL. Since studies have shown that no additional protection is achieved with trough IgG levels higher than 400 mg/dL, the six-month result indicates that optimal therapy has been achieved. IgM and IgA levels remain low because the IVIG preparation does not contain significant amounts of these immunoglobulins. One year later, Sam shows definite clinical improvement; he has been without infections since IVIG therapy was started.
The differential diagnosis of Sam's immunodeficiency starts with the immunologic studies conducted above that show the presence of a B-cell defect. However, even before such data are obtained, certain features of Sam's clinical course allow informed speculation concerning his diagnosis.
What are these features?
In evaluating where Sam's defect lies, it is helpful to look at a scheme of B-cell development and function and consider where the block may occur (Fig. 1.1).
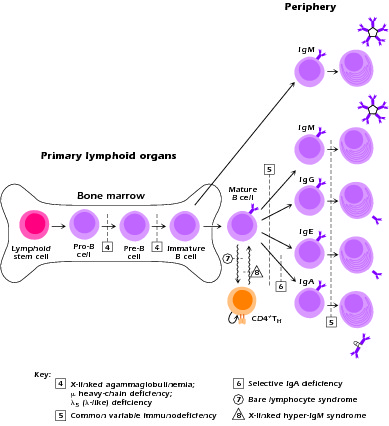
Figure 1.1 Scheme of B-cell development and function and locations of blocks in development with various immunodeficiency disorders.
How does Sam's workup allow one to pinpoint the problem?
Correct answer:
Sam's immunologic workup revealed a reduction in all the serum immunoglobulins and an absence of response to immunization. The absence of all immunoglobulin isotypes suggests that the block occurs at a stage prior to the development of isotype-specific plasma cells. Specifically, this eliminates a selective IgA deficiency and hyper-IgM syndrome. The latter is unlikely because Sam's IgM levels are low and because some forms of hyper-IgM syndrome result from T-cell abnormalities.
Working our way to the left in Figure 1.1, we encounter common variable immunodeficiency (CVID) and bare lymphocyte syndromes. Bare lymphocyte syndrome, in which the cells lack the expression of one of the human leukocyte antigen (HLA) classes, would lead to a defect in DTH and humoral immunity combined, since both depend on class II MHC expression. Lack of class I MHC antigen expression would affect T cytotoxic activity and spare T helper cell stimulation.
It is somewhat more difficult to clinically distinguish between CVID and X-linked agammaglobulinemia (XLA), which might both be part of your differential diagnoses. Like XLA, CVID is dominated by a humoral immune defect manifested by the inability to mount normal antibody responses, and the two have similar infectious histories. However, CVID usually manifests later than XLA and indeed was initially called "acquired agammaglobulinemia" because it has its onset in adulthood more often than in childhood. Four years of age would be particularly young for CVID. In addition, patients with CVID not infrequently manifest various types of autoimmune manifestations leading to chronic gastrointestinal (GI) disorders, hematopoietic abnormalities, and liver abnormalities that are never seen in XLA. Importantly, in CVID, mature B cells are present, although the activation and differentiation to antibody-producing cells are blocked. In Sam, the defect is earlier and prevents the development of mature B cells. As you will note, we have worked our way back to the earliest stages of B-cell development to localize the point of Sam's block (Fig. 1.2).
A final differential must be made among the various types of B-cell abnormalities that result in a block in early B-cell development at the pro-B cell stage and thus lead to the absence of mature B cells in the periphery: XLA, μ heavy chain deficiency, and λ5 deficiency (see the section on normal B-cell development below for events leading to B-cell maturation). The distinction among these diseases on clinical or even on immunologic grounds can be quite difficult. However, both μ heavy chain deficiency and λ5 chain deficiency show autosomal recessive patterns of inheritance and are quite rare. The family history of healthy females and affected male relatives on Sam's mother's side is classic for an X-linked Mendelian genetic disease and is thus more consistent with XLA. Another difference is that μ heavy chain deficient patients generally display even lower levels of IgG than XLA patients and have undetectable peripheral B cells. Both XLA and λ5 deficiency patients generally have very small numbers of circulating B cells, but in XLA the B cells have an immature phenotype, and in λ5 deficiency they have a mature phenotype.
In the end, distinction among these early B-cell defects requires confirmation by molecular analysis. This is usually done by analysis of patient monocytes, another cell that ordinarily expresses Bruton's tyrosine kinase (Btk) (although lack of Btk in monocytes does not cause abnormalities of monocyte function in XLA). Identification of the mutation may be necessary for screening of future pregnancies for the occurrence of affected fetuses.
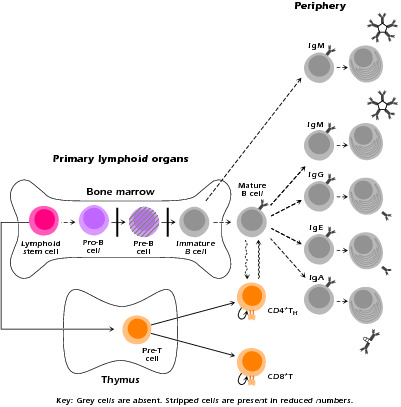
Figure 1.2 Remaining developing and mature lymphoid populations in XLA. Gray cells are populations missing in XLA patients. Bars indicate positions of developmental blocks.
XLA is characterized by susceptibility to infection with pyogenic bacterial organisms, particularly encapsulated Gram-positive and Gram-negative organisms such as Haemophilus influenzae, S. pneumoniae, S. aureus, and Pseudomonas species. It thus results in severe recurrent acute infections starting at approximately six months of age when maternal antibodies transmitted to the child via the placenta in utero are no longer present in sufficient amount. Upper and lower respiratory infections are especially common, but life-threatening sepsis and meningitis (as in the present case) also occur. A particularly serious and long-term problem is recurrent pneumonia because it can lead to bronchiectasis, pulmonary fibrosis, and pulmonary failure. As previously noted, a clue to the early diagnosis and treatment of XLA (and a clinical pearl) is the absence of lymph node enlargement and underdeveloped tonsils in a young child who presents with what seem to be normal childhood infections.
Organisms that enter the bloodstream through the GI tract are a particular problem for patients with B-cell deficiencies. Thus, immunization with live poliovirus may lead to infection rather than protection. This was the probable scenario in Sam's mother's cousin who died of poliomyelitis following "vaccination." A particularly ominous type of infection that can occur in XLA (as well as in CVID) is enteroviral infection of the central nervous system (CNS). This infection can lead to a poorly understood type of autoimmune encephalomyelitis that results in progressive loss of CNS function characterized by motor and intellectual deterioriation and culminating in death. It is refractory to treatment since it is usually recognized when the infectious phase is over and the CNS is sterile. In some cases XLA is associated with a dermatomyositis-type disorder. As previously mentioned, in sharp contrast to CVID patients, XLA patients do not manifest the array of autoimmune disorders mentioned above. Other viral and fungal infections are well handled by XLA patients.
In the fetus, B-cell development first begins, along with all hematopoiesis, in the liver and then transfers to the bone marrow, the major site of development at birth. The orderly process of hematopoiesis is dependent on the interaction of precursor cells with their microenvironment, such as, bone marrow stromal cells and osteoblasts, in ways which are still being elucidated. In order to produce mature B cells, the first decision point is at the level of a pleuripotential hematopoietic stem cell in the bone marrow which commits to the lymphoid lineage. The next decision point is the determination as to whether differentiation will progress along the T/NK pathway or the B-cell pathway. We will pick up the story of B-cell development at the level of the progenitor B cell (pro-B), as illustrated in Figure 1.3. Note that most of the experimental data have been obtained on mice, and there are differences between human and mouse B-cell development. The nomenclature also varies (even from investigator to investigator) within one species. We will focus on human data where available.
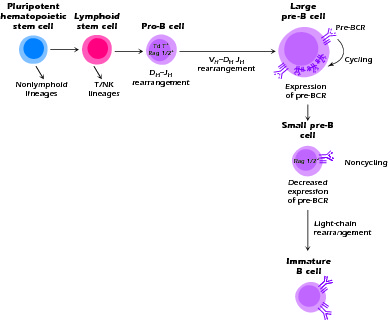
Figure 1.3 B-cell development in the bone marrow. Patients with XLA do not expand their pre-B cell population due to lack of signaling and the cells do not survive.
During normal B-cell development, the pro-B cell initiates immunoglobulin synthesis with the rearrangement and transcription of the μ heavy-chain gene. This occurs first with the joining of the DHJH segments, followed by the joining of VH segments. Accomplishing this task requires the expression of a variety of transcription factors and recombination enzymes (Rag 1 and 2), endonucleases, and TdT (terminal deoxynucleotidyl transferase) similar to those operating in the developing T cell. The productive rearrangement of the μ heavy-chain gene culminates in the expression of μ protein on the cell surface plasma membrane and/or in the internal cytoplasmic membranes in association with a "surrogate" light chain. The surrogate light chain is the product of two genes, VpreB and λ5 (also called λ-like in humans). The heavy chains and surrogate light chains assemble into an Ig-like complex and become associated with two transmembrane signal-transducing molecules, Igα and Igβ (CD79a and CD79b). The molecular complex thus formed is called the pre-B-cell receptor (pre-BCR) and the cell is now termed a precursor (pre)-B cell. The down-regulation of the recombination enzymes, further differentiation, continued survival, and, in humans, the proliferation of the pre-B cell requires stimulation via the pre-BCR and the activation of a pre-BCR-dependent signaling cascade. This includes the activation of a key cytosolic tyrosine kinase enzyme known as Bruton's tyrosine kinase, which is a Tec family protein tyrosine kinase (PTK) (Fig. 1.4).
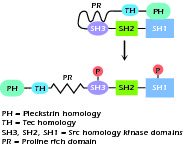
Figure 1.4 Structure of Bruton's tyrosine kinase, a Tec family protein tyrosine kinase. Upon activation by phosphorylation molecule unwinds into a linear strand.
The activation of Btk and its role in B-cell development is illustrated in Figure 1.5. A description of this biochemical pathway is as follows:
Activation, possibly by cross-linking, of the pre-BCR causes phosphorylation of the cytoplasmic tails of Igα and Igβ on their ITAM domain (immunoreceptor tyrosine-based activation motif). These then activate the Src family kinases, particularly Lyn and then Syk and Blk. This allows the formation of a docking station for Syk, which then recruits and phosphorylates SLP-65 and Btk at its PH (Pleckstrin homology) domain. The adaptor protein SLP-65 (also known as BLNK or BASH) functions as a scaffold, bringing Btk to the membrane. This activated SLP-65 molecule further recruits Btk and phospholipase Cγ2 (PLC-γ2). Btk activates PLC-γ2, which then hydrolyzes PIP2 (phophatidylinositol bisphosphate) to IP3 (inositol triphosphate) and DAG (diacylglycerol). This opens calcium channels and activates protein kinase C. In humans, SLP-65 and Btk control both differentiation and proliferation of the pre-B cell, whereas in mice, although differentiation appears dependent on these molecules, proliferation involves different pathways and appears dependent on IL-7 signaling. Proliferation involves the activation of the RAS and MAPK (mitogen-activated protein kinase) pathways. Thus in humans, mutations in Btk and the much more rare mutations in SLP-65 or λ5 result in a severe phenotype; mice with mutations in these molecules or "knockouts" demonstrate milder and "leakier" B-cell deficiency (see Unit V for description of knockout mice). Recent evidence suggests that, after activation, Btk and SLP-65 down-regulate the pre-BCR, thereby limiting pre-B-cell proliferation and possibly functioning as tumor suppressor molecules.
Activated Btk again functions in the mature B cell and may integrate pathways important for its survival, proliferative response, and modulation of the signal threshold necessary for B-cell activation via the BCR. In addition, Btk is expressed in mast cells and may play a role in their ability to degranulate in response to surface receptor cross-linking. Btk expression is restricted to the B-cell lineage, myeloid (granulocytic) cells, and erythroid cells. Finally, Btk is expressed by and is necessary for the proliferation of leukemic cells in patients with chronic lymphocytic leukemia; thus, it is a treatment target in this disease.
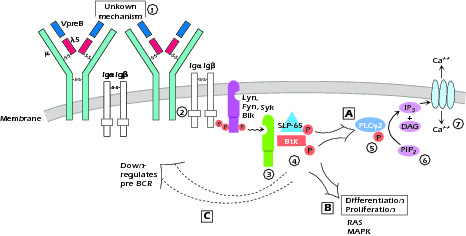
Figure 1.5 The activation of Btk and its role in B-cell development 1 Activation of pre-BCR by unknown mechanism (? crosslinking by ligand) brings 2 Src family kinases, Lyn, Fyn and Blk to the ITAMS on Igα and Igβ as a result of their phosphorylation on their ITAMS. This results in 3 a docking site for Syk which 4 recruits and phosphorylates SLP-65 and Btk. 5 BtK in turn associates with and activates PLCγ. 6 Activated PLCγ2 hydrolyzes PIP2 to DAG + IP3 7 which opens Ca++ channels in the membrane. SLP-65 and BtK also activate differentiation and proliferation, the latter through RAS and MAPK pathways and then down-regulate the pre-BCR. A, B, and C denote the three pathways affected by SLP-65 - Btk activation.
The pre-B cell has now gone from a larger proliferating cell to a small pre-B cell. It has exited the cell cycle and down-regulated pre-BCR expression and is no longer synthesizing surrogate light chain molecules. Re-expression of the enzymes required for immunoglobulin gene rearrangement, Rag 1 and 2, initiates light chain gene rearrangement and protein synthesis. This light chain must be able to associate with the μ heavy chain on the surface for the cell to escape apoptosis. The cell is now the immature B cell and is subject to negative selection for elimination of autoreactive cells.
X-linked agammaglobulinemia, or Bruton's agammaglobulinemia, was described by Ogden Bruton in 1952 and, indeed, was the first description of an immunodeficiency disorder. Dr. Bruton, a colonel in the U.S. army at Walter Reed Hospital in Washington DC, identified the first case when he noticed that a patient with recurrent infections had a very low gamma globulin band in the recently developed protein electrophoresis test. The disease has a frequency of 3-6 per million. It was not until 1993 that the responsible protein was identified and its gene mapped to the long arm of the X chromosome at Xq21.2-22. It was subsequently named for Dr. Bruton, Bruton's tyrosine kinase.
In the presence of a mutated Btk and the consequent absence of signaling of the pre-B cell via the pre-BCR, the pre-B cell undergoes apoptosis or fails to mature. The B lineage cells thus accumulate in the marrow at the pro-B cell stage with few pre-B cells identifiable. Instead of the larger cycling cells normally seen at the beginning of the pre-B cell stage, those pre-B cells that are present are small. This block in proliferation and differentiation leads to an absence of mature B cells in the periphery; no or few circulating immature B cells; tonsils and lymph nodes that lack germinal centers; and the absence of plasma cells. Recently it has been shown that bone marrow B cells in XLA patients do produce antibodies, but these antibodies have altered reactivity, leading in some cases to autoantibody activity. Thus, intact Btk function may also be necessary for deletion of B cells that react to autoantigens.
Over 300 different mutations that give rise to XLA have been identified in the BTK gene. These range from point mutations with single amino acid substitutions to deletions and stop codons. It should be noted that XLA is one of several immunodeficiency disorders caused by a mutation in a tyrosine kinase enzyme. The other two thus far identified are mutations in JAK3, which causes a form of SCID, and mutations in ZAP70, which results in a type of T-cell deficiency.
Several other genetic abnormalities, μ chain deficiency, λ5 (also called λ-like in humans) deficiency, and Iga deficiency are similar to XLA in that they also lead to a defect in BCR expression at the pro/pre-B-cell stage of B-cell development. This emphasizes the fact that expression of the pre-BCR and its signaling are necessary for maturation of the B cell beyond the pro/pre-B-cell stage. These autosomal recessive abnormalities, which have been described in only a handful of patients, are clinically similar to XLA, although μ chain deficiency is a somewhat more severe disease with an earlier onset and a more profound lack of Ig. The exclusive accumulation of pro/pre-B cells in the bone marrow of patients with these genetic abnormalities allows definition of its phenotype as CD19+CD34+ B cells, defines the bone marrow as the site of B-cell maturation in nonavian species (birds have the bursa of Fabricius for maturation of B cells), and suggests the existence of little or no extramedullary B-cell maturation. Recall that the mouse model for XLA (the xid mouse), which is also due to a mutation, is characterized by much less severe symptomatology, suggesting that in murine B cells, non-Btk signaling can partially compensate for Btk signaling or that Btk functions are more restricted in mice.
What would you observe in a female carrier of XLA who is heterozygous for glucose-6-phosphate dehydrogenase (G6PD) isozyme?
XLA and related B-cell deficiencies are logically and effectively treated by the administration of IVIG. Such treatment usually prevents serious lower respiratory infection and thus prevents the development of bronchiectasis and other chronic pulmonary problems. IVIG therapy has supplanted intramuscular IG therapy because it results in higher levels of serum IgG and because it avoids the local skin and muscle problems associated with the injection of intramuscular injection of IG. IVIG contains only low amounts of IgM and IgA, and thus IVIG therapy does not restore the levels of these immunoglobulins. In addition, IgA normally functions at the mucosal surface and would not be effectively if replaced in this manner. If infection occurs in spite of IVIG therapy, it is important to treat the patient early with appropriate antibiotics. As with any genetic disorder resulting from the absence of a single protein, gene replacement therapy is the hope of the future.
As these patients live longer, the possibility of an increased incidence of malignancies, particularly hematologic malignancies, arises. Interestingly, gastric carcinoma, also frequent in CVID patients, is reported at a higher than expected rate in XLA patients.